


Dépistage néonatal : cinquante ans de progrès
Développé dès les années 1950 aux États-Unis, le dépistage néonatal s’est rapidement étendu à d’autres pays. Lancé en 1972 dans l’hexagone, le programme de dépistage néonatal à la française a vu l’ajout de sept nouvelles pathologies rares en 2023. Retour sur ces décennies de recherche et d’avancées pédiatriques.
1951 : Les pionniers américains
Initiés outre-Atlantique dès la fin des années 1940, les programmes de recherche sur les dépistages néonataux se sont répandus au cours des décennies suivantes à travers le monde. Le premier événement majeur sur le sujet est la Commission sur les maladies chroniques (Commission on Chronic Illness, en anglais) en 1951.
Elle a donné une définition claire des enjeux : « Le dépistage consiste à identifier présomptivement à l’aide de tests, d’examens ou d’autres techniques susceptibles d’une application rapide, des sujets suspects d’être atteints d’une maladie ou d’une anomalie passée jusque-là inaperçue. »
1963 : Robert Guthrie, père du dépistage moderne
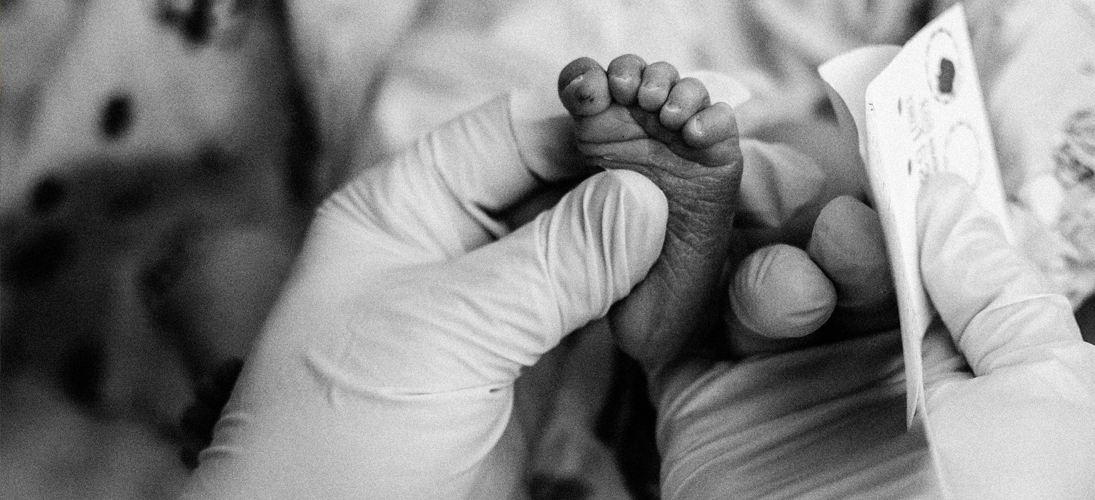
L’histoire du dépistage néonatal bascule en 1963 lorsque le médecin et microbiologiste américain Robert Guthrie met au point un test qui porte aujourd’hui son nom. Celui-ci s’effectue entre la 72e et la 96e heure de vie du nourrisson et consiste en une simple piqûre au niveau du talon ou à l’arrière de la main du nourrisson. Six gouttes de sang sont prélevées. Elles sont ensuite disposées sur un papier-filtre (d’où le surnom de « test du buvard ») et analysées en laboratoire. Les résultats sont connus entre 24 et 48 heures après. La simplicité de la procédure ouvre la possibilité d’un dépistage de masse.
Le médecin américain s’est intéressé de près à la question des dépistages et à la pédiatrie du fait de son histoire personnelle : son deuxième enfant est né avec un retard mental et sa nièce est atteinte de phénylcéturonie. Cette pathologie peut également entraîner un retard mental sévère. Elle est due à l’incapacité de l’organisme à dégrader un acide aminé essentiel pour l’organisme, appelé la phénylalanine, qui s’accumumle dans le sang et devient alors toxique pour le cerveau. Cependant, cette maladie génétique peut être traitée lorsqu’elle est prise en charge à temps.
1966 : La France s’empare du sujet
En France, il faut attendre 1966 pour que des études de faisabilité du test de Guthrie soient menées dans divers centres hospitaliers, notamment à Lyon et à Lille. L’année suivante, la promotion du test du buvard est assurée par un acteur plutôt surprenant. C’est en effet la Société des eaux d’Évian qui s’empare de la problématique et met à disposition des chercheurs un de ses laboratoires pour réaliser des dépistages de la phénylcétonurie. À la fois mécène et aide logistique, l’entreprise propose aussi un test gratuit aux maternités du secteur.
Une activité novatrice que ce laboratoire savoyard assure seul jusqu’en 1972, lorsque la Sécurité sociale prend le relais et rembourse intégralement les dépistages, étendant la possibilité d’un test à toute la France. La création cette même année de l’Association française pour le dépistage des handicaps de l’enfant (AFDPHE) vient consolider le programme français. En 1979, les tests deviennent disponibles pour les territoires et départements d’outre-mer.
1968 : L’OMS donne un cadre
Cette année-là, l’Organisation mondiale de la santé (OMS) commande un rapport sur la question des dépistages à James Wilson, médecin en chef au ministère de la Santé d’Angleterre, et à Gunner Jungner, alors chef du département de chimie clinique de l’hôpital de Göteborg, en Suède. Intitulé Principes et pratiques du dépistage des maladies, mais souvent désigné sous le nom de « rapport Wilson et Jungner », ce document acte les enjeux et bonnes pratiques de dépistage. Dix points sont édictés par les deux chercheurs notamment sur l’importance d’avoir déjà développé un « traitement d’efficacité démontrée » à la maladie à dépister, sur la capacité à la déceler « pendant une phase de latence ou au début de la phase clinique » et aussi qu’elle constitue « une menace grave pour la santé publique ».
2023 : Sept nouvelles pathologies en France
Si le dispositif de dépistage s’est perfectionné et pérennisé au fil des décennies, la France accusait, jusqu’à récemment, un certain retard par rapport à ses voisins européens avec seulement six maladies dépistées. La Suède et l’Autriche dépistent 24 maladies, la plupart des pays d’Europe de l’Ouest au moins 15. Depuis 2020, la Haute Autorité de santé (HAS) prônait l’intégration de sept nouvelles pathologies au test : la tyrosinémie de type 1, l’acidurie glutarique de type 1, l’acidurie isovalérique, la leucinose, l’homocystinurie, le déficit de captation de la carnitine et le déficit en déshydrogénase des hydroxyacyl COA à chaîne longue. Ces recommandations ont finalement été suivies par le ministère de la Santé qui les a intégrées le 1er janvier 2023. Tous les nouveau-nés bénéficient désormais de 13 dépistages, avec l’accord des parents.
Sources : Université de Lorraine ; Haute Autorité de santé (HAS) ; Medecinesciences.org ; Bibliothèque nationale de médecine (NIH) ; CHU de Lyon ; Santé publique France.
© C i E M / Mathieu Yerle
Quelles sont les 13 maladies dépistées ?
Désormais, ces maladies rares, sévères et le plus souvent génétiques sont dépistées à la naissance :
- la phénylcétonurie :un déficit de l’enzyme qui transforme la phénylalanine, un acide aminé essentiel pour l’organisme ;
- l’hypothyroïdie congénitale : une sécrétion insuffisante des hormones thyroïdiennes ;
- l’hyperplasie congénitale des surrénales : un défaut de fonctionnement des glandes surrénales ;
- la mucoviscidose : un épaississement des sécrétions des poumons et du pancréas ;
- le déficit en MCAD (Medium-chain-acyl-CoA déshydrogénase) :un déficit qui empêche de transformer les graisses en énergie ;
- l’homocystinurie : un déficit qui entraîne l’accumulation d’un acide aminé, l’homocystéine, toxique pour l’organisme ;
- la leucinose : le déficit d’une enzyme qui intervient dans la transformation d’acides aminés ;
- la tyrosinémie de type 1 : un déficit de l’enzyme qui permet la transformation des protéines ;
- l’acidurie isovalérique : un déficit d’une enzyme entraînant la formation de substances toxiques ;
- l’acidurie glutarique de type 1 : le dysfonctionnement d’une enzyme qui intervient dans la dégradation d’acides aminés ;
- le déficit en déshydrogénase des hydroxyacyl COA à chaîne longue : un déficit qui provoque l’accumulation d’acides gras ;
- le déficit de captation de la carnitine : un déficit qui empêche l’organisme de décomposer correctement les graisses ; ν la drépanocytose : la présence d’une hémoglobine anormale dans le sang.
Ce programme est complété par le dépistage de la surdité permanente néonatale.

